FDA regulated research
FDA regulated research refers to human subjects research that involves the use of medical devices, drugs, or biological products, that are regulated by the U.S. Food and Drug Administration (FDA). When a study involves a product that is intended to diagnose, cure, mitigate, treat, or prevent disease, or that affects the structure or function of the human body, FDA regulations may apply.
Unlike human subjects research governed solely by the Common Rule (45 CFR 46), the FDA uses the term “clinical investigation” to describe research involving FDA‑regulated products. A clinical investigation is any experiment involving one or more human subjects and a test article (such as a drug or device) where the results are intended to be submitted to, or held for inspection by, the FDA.
Studies that are FDA regulated must comply with both IRB requirements and applicable FDA regulations, including requirements related to informed consent, safety monitoring, and investigational product oversight.
What is a medical device?
A medical device is an instrument, apparatus, implement, machine, implant, or similar test article intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease, or intended to affect the structure or function of the body. Examples include Diagnostic, Investigational, In-Vitro, and Software as Medical Device. Medical devices do not achieve their primary intended purposes through chemical action or by being metabolized.
Medical devices used in research are regulated by the FDA and are classified based on the level of risk they pose to participants.
The FDA classifies medical devices into three categories: Class I, Class II, and Class III. The classification determines the level of regulatory oversight required.
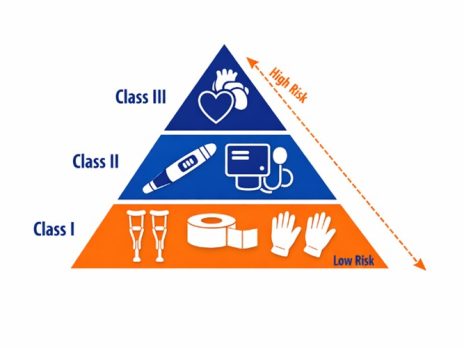
Class I devices (low risk)
Class I devices pose minimal potential risk to participants and are subject only to General Controls. These controls include requirements for registration, labeling, and good manufacturing practices.
Examples may include:
- Simple surgical instruments
- Bandages, thermometers
- Mouthpieces
Most Class I devices are exempt from premarket submission requirements.
Class II devices (moderate risk)
Class II devices pose a moderate level of risk and are subject to General Controls and Special Controls. Special controls may include performance standards, post‑market surveillance, and specific labeling requirements.
Examples may include:
- Infusion pumps
- Blood pressure cuffs
- Diagnostic imaging accessories
Most Class II devices require FDA clearance through a 510(k) premarket notification, unless exempt.
Class III devices (high risk)
Class III devices pose the highest level of risk and are typically life‑supporting, life‑sustaining, implanted, or otherwise of substantial importance in preventing impairment of human health.
Examples may include:
- Implantable pacemakers
- Heart valves
- Certain neural or vascular implants
Class III devices generally require premarket approval (PMA) from the FDA and often involve an investigational device exemption (IDE) when used in research.
FDA IDE and exemptions
An IDE is required when conducting a clinical investigation of a medical device that is not deemed IDE exempt or not a significant risk device (NSR). IDEs are required when the clinical investigation of the medical device is a significant risk (SR).
The FDA recognizes certain categories of device research that do not require IDE approval, even though the device is being used in a clinical investigation.
If the device meets IDE exempt criteria or meets the definition of not significant risk (NSR), an IDE is not required.
An NSR device study is one that does not meet the definition for an SR device study.
What is a drug in research?
In FDA regulated research, a drug includes any article (other than food) intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease and articles intended to affect the structure or any function of the body.
This includes:
- Investigational drugs
- Approved drugs used for a new indication, dosage, route of administration, or population
- Certain biologics and vaccines
Clinical investigations involving drugs may require an investigational new drug (IND) application unless the study qualifies for an exemption under FDA regulations.
Investigational new drug (IND) and exemption considerations
An IND provided by the FDA allows an investigational drug to be administered to humans in a clinical investigation. Even when an IND is held by a sponsor or collaborator, or the study qualifies for exemption, IRB review and approval are still required prior to initiating the research.
Biological product (biologic)
The FDA defines a biologic as a virus, therapeutic serum, toxin, antitoxin, vaccine, blood, blood component or derivative, allergenic product, protein (except any chemically synthesized polypeptide), or analogous product applicable to the prevention, treatment, or cure of a disease or condition of human beings.
Common examples of biologics
- Vaccines
- Gene therapies
- Allergen extracts
- Monoclonal antibodies
Resources and guidance
FDA contacts for devices, drugs and biologics
For product‑specific questions, researchers and sponsors may contact the FDA directly.
- General FDA Contact:
1‑888‑INFO‑FDA (1‑888‑463‑6332)
https://www.fda.gov/about-fda/contact-fda - Medical Devices (CDRH):
https://www.fda.gov/medical-devices - Drugs and Biological Products (CDER/CBER):
https://www.fda.gov/drugs
FDA Q‑Submission program (devices)
The FDA Q‑Submission (Q‑Sub) program allows researchers and sponsors to request feedback or meetings with the FDA prior to or during the development of a medical device. This program is commonly used to clarify regulatory expectations before submitting an IDE, 510(k), or PMA.
FDA Product Classification Database
FDA Guidance – Significant Risk and Nonsignificant Risk Medical Device Studies
NIH Device Study Risk Determination & IDE Exemption Criteria Decision Tree
Need help determining FDA applicability?
If you are unsure whether your research is FDA regulated, or whether an IND or IDE is required, contact the NAU IRB/HRPP office early in the study planning process. Early consultation can help prevent delays and ensure regulatory compliance.
Version 2026-4